

Research Article
Volume-1 Issue-1, 2026
Study of Hydrogen Transfer Reactions within the Framework of the Non-Equilibrium Approach; CH4 + OH → CH3 + H2O Reaction
Received Date: January 05, 2026
Accepted Date: January 20, 2026
Published Date: January 23, 2026
Journal Information
Switch to Full Text Menu
Abstract
A theoretical study of the gas-phase reaction CH4 + OH → CH3 + H2O kinetics (energy calculation level CCSD (T)/6-311++G**//B3LYP/6-31+G**) in the temperature range 200 – 2000 K was performed. A discussion is held within the framework of the non-equilibrium model, in which at the fixed C.O distance the tunneling H-atom and system reorganization processes occur simultaneously. Kinetic analysis, performed with regard to a promoting effect of C (H).O bond oscillation and the lifetime of the collision complex, leads to a close agreement with experimental data.
Key words
Methane, Hydroxyl Radical, Non-Equilibrium Model, Promoting Vibrations, Collision Complex Lifetime
| Parameter | This worka) | [4]b |
| ΔE00 | 10.91 | -13.3 |
| ESP | 8.52 | 6.4 |
| Q, A | Ea | Eb1 | Eb2 | rb, А |
| 2.5 | 4.216 | 4.331 | 11.225 | 1.205 |
| 2.6 | 2.805 | 6.572 | 15.15 | 1.29 |
| 2.7 | 1.823 | 10.168 | 19.979 | 1.36 |
| 2.8 | 1.146 | 14.634 | 25.336 | 1.419 |
| 2.9 | 0.675 | 19.708 | 31.021 | 1.479 |
| 3 | 0.339 | 25.165 | 36.866 | 1.537 |
| Q,A | r01 | rb | r02 |
| 2.5 | 113.6 | 174 | 160.6 |
| 2.6 | 119.4 | 177.2 | 163.2 |
| 2.7 | 125 | 177.9 | 165.1 |
| 2.8 | 130.7 | 178.4 | 166.5 |
| 2.9 | 136.3 | 178.5 | 167.7 |
| 3 | 141.9 | 178.6 | 168.2 |
| Q, A | τt′ , s | η | τt , s | ν(CH3), cm-1 | T0.5, s | n |
| 2.6 | 4.2(-15) | 11.5 | 4.85(-14) | 1314 | 1.27(-14) | 3.82 |
| T, K | Theory | Experiment | |||||||
| k | k0 | k1 | k2 | k3 | k4 | k1′ | [1] | [2] | |
| 2000 | 1.02(10) | 1.43(10) | 3.84(10) | 6.32(10) | 9.27(10) | 1.17(11) | 8.27(9) | 8.73(9) | 1.09(10) |
| 1500 | 3.10(9) | 4.60(9) | 1.32(10) | 2.24(10) | 3.84(10) | 4.35(10) | 3.45(9) | 3.86(9) | 4.66(9) |
| 1000 | 5.01(8) | 8.29(8) | 2.59(9) | 4.53(9) | 7.14(9) | 9.31(9) | 8.85(8) | 1.08(9) | 1.23(9) |
| 800 | 1.70(8) | 3.03(8) | 9.78(8) | 1.74(9) | 2.79(9) | 3.72(9) | 3.88(8) | 4.94(8) | 5.39(8) |
| 600 | 4.48(7) | 8.77(7) | 2.85(8) | 5.15(8) | 8.26(8) | 1.11(9) | 1.37(8) | 1.60(8) | 1.64(8) |
| 500 | 1.75(7) | 3.65(7) | 1.18(8) | 2.15(8) | 3.43(8) | 4.63(8) | 6.40(7) | 7.22(7) | 7.03(7) |
| 400 | 5.00(6) | 1.13(7) | 3.59(7) | 6.54(7) | 1.03(8) | 1.40(8) | 2.26(7) | 2.43(7) | 2.20(7) |
| 350 | 2.25(6) | 5.36(6) | 1.66(7) | 3.02(7) | 4.73(7) | 6.37(7) | 1.15(7) | 1.17(7) | 1.02(7) |
| 300 | 8.54(5) | 2.16(6) | 6.49(6) | 1.17(7) | 1.80(7) | 2.40(7) | 4.95(6) | 4.71(6) | 3.81(6) |
| 250 | 2.46(5) | 6.71(5) | 1.92(6) | 3.41(6) | 5.13(6) | 6.74(6) | 1.66(6) | 1.41(6) | 1.04(6) |
| 200 | 3.98(4) | 1.23(5) | 3.31(5) | 5.76(5) | 8.43(5) | 1.08(6) | 3.31(5) | 2.56(5) | 1.66(5) |
| Q, A | E0 | Tcol 10-13, s | |
| cm-1 | kcal.mol-1 | ||
| 2.5 | 304.9 | 0.436 | 1.09 |
| 2.6 | 255.9 | 0.365 | 1.3 |
| 2.7 | 213.8 | 0.306 | 1.56 |
| 2.8 | 177.5 | 0.254 | 1.88 |
| 2.9 | 148.4 | 0.212 | 2.25 |
| 3 | 132 | 0.188 | 2.52 |
| j | Ej, kcal.mol-1 | Qj, A | νt(Qj) | fj a) | Pj b) | Fj(Q) | F(Q) | kpr(Q) |
| 0 | 0.254 | 2.759 | 2.47(10) | 6.7 | 1 | 6.7 | 6.7 | 1.764(6) |
| 1 | 0.762 | 2.661 | 1.06(12) | 287 | 0.464 | 133.3 | 140 | 3.688(7) |
| 2 | 1.27 | 2.611 | 4.58(12) | 1244 | 0.279 | 346.6 | 487 | 1.282(8) |
| 3 | 1.778 | 2.57 | 1.19(13) | 3235 | 0.167 | 540.4 | 1027 | 2.706(8) |
| 4 | 2.286 | 2.534 | 2.24(13) | 6094 | 0.1 | 610.6 | 1638 | 4.314(8) |
| σ ,cm | ε,cm-1 | |
| CH4 | 3.41 | 14.9 |
| OH | 2.66 | 19.9 |
| H3C(H)…OH | 2.86 | 17.22 |
| T, K | τlf 10-13, s | р | f | T, K | τlf 10-13, s | р | f |
| 2000 | 0.97 | 0.75 | 0.215 | 400 | 2.85 | 2.19 | 0.63 |
| 1500 | 1.18 | 0.91 | 0.261 | 350 | 3.12 | 2.4 | 0.689 |
| 1000 | 1.55 | 1.19 | 0.342 | 300 | 3.45 | 2.66 | 0.763 |
| 800 | 1.8 | 1.38 | 0.397 | 250 | 3.9 | 3 | 0.862 |
| 600 | 2.18 | 1.67 | 0.481 | 200 | 4.52 | 3.48 | 1 |
| 500 | 2.46 | 1.89 | 0.543 |
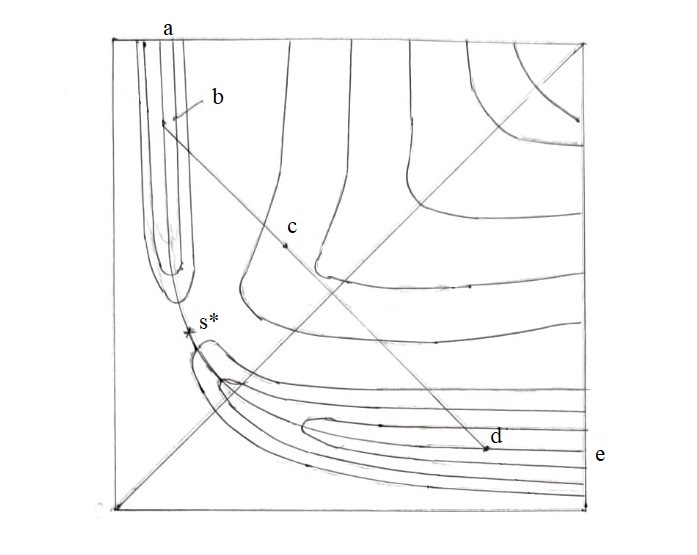 |
| Figure 1: The route of the system in the non-equilibrium reaction of H-atom transfer. Comments in the text. |
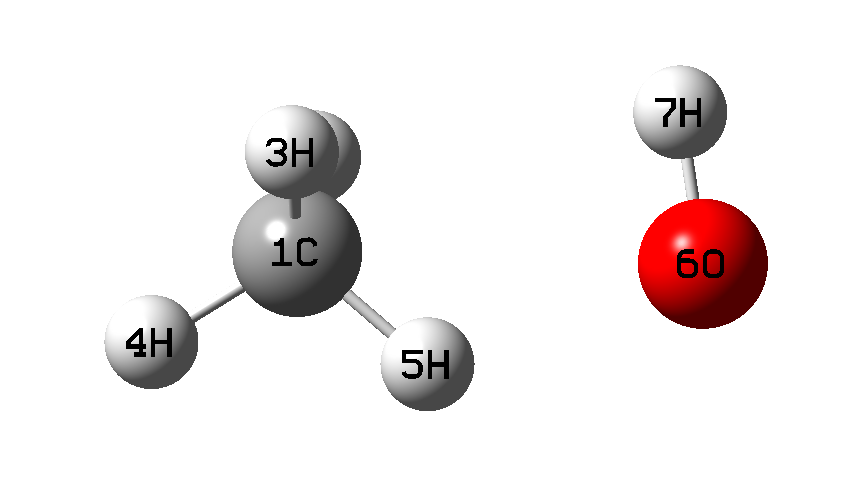 |
| Scheme 1 |
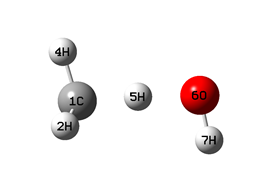 |
| Scheme 2 |
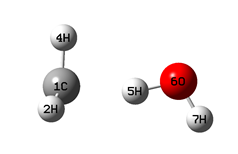 |
| Scheme 3 |
 |
 |
| Figure 3: The influence of C...O vibrations in the collision complex on the system route in the reaction CH4 + OH. |
 |
| Figure 4: Reaction rate constant kpr(Q) (Eq. 16) at T = 500 K as a function of C…O bond promoting vibration levels (curve number in parentheses): zero (2), zero and first (3), zero – second (4), zero – third (5), zero – fourth (6); k(Q) in Eq. 7 (1). |
 |
| Figure 5: Thermal rate constant for reaction СН4 + ОН: calculation using Eq. 21 (curve 1); experiment: [1] (curve 2) and [2] (curve 3). |
Introduction
The reaction of methane with the hydroxyl radical (Eq. 1) has attracted the attention of researchers for decades due to the key role this reaction plays both in the balance of atmospheric methane and in hydrocarbon combustion processes.
CH4 + OH → CH3 + H2O (1)
As a result of intensive experimental studies of the reaction, performed by different scientific groups in a wide range of temperatures, reliable kinetic data were obtained, characterized by mutual consistency: the equations recommended in the reviews of 1986 [1] (Eq. 2) and 2005 [2] (Eq. 3) lead to very close values of the rate constants.
k (T) = 2.49.10-18 T2.13 exp(-1230/T) 250 -2000 K (2)
k (T) = 2.27.10-18 T2.18 exp(1350/T) 250 -2400 K (3)
in cm3 molecule-1 s-1
Recent theoretical studies have been aimed at high-level calculation of the full-size PES of the reaction [3-5] and description of its kinetics both regard to the variational TST with correction for tunneling [6, 7] and using dynamic modeling [4, 6, 8-10] (only a few references are given).
The present work continues the study of gas-phase reactions of hydrogen atom transfer within the framework of the non-equilibrium approach. Previously, an attempt was made to use the data on the kinetics of reactions of the methyl radical with methane [11] and methanol (along the C–H [12] and O–H [13] bonds) to describe hydrogen atom tunneling dynamic aspects [13, 14]. The present work is devoted to examining the system dynamics at the moment of reactant collision from this point of view.
Theory
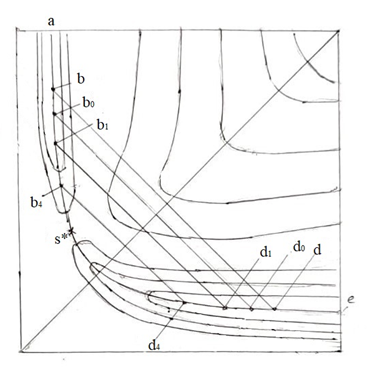
The non-equilibrium theory of proton transfer in solution [15-18] is based on the generalized Franck-Condon principle (GFCP) [19]: during proton tunneling, heavy atoms of the system retain their positions. As shown by the study of the reactions of the methyl radical with methane (Eq. 4) and methanol (Eqs. 5, 6), for H-atom transfer reactions in the gas phase, the factor of constancy of the distance between the H-donor and acceptor atoms, Q, during tunneling comes to the fore [13]. In the potential energy diagram (Figure. 1), this condition determines the motion of the reaction system along the abde route; sections ab and de correspond to the motion of the sys-tem along the minimum energy path (MEP) in the reactant and product valleys, respectively, and section bd corresponds to the tunneling of the H-atom; at a fixed distance Q (in the figure distance bd), tunneling occurs in a double-well potential V(Q; r), where r is the tunneling coordinate .
CH4 + CH3 → CH3 + CH4 (4)
CH3OH + CH3 → CH2OH + CH4 (5)
CH3OH + CH3 → CH3O + CH4 (6)
It is easy to show the difference between this method of the reaction description and that one adopted in the classical TST and modern dynamic models. Let us imagine that at point b (Figure. 1) the system has some excess kinetic energy. If this is the translational energy of the relative motion of the reactants, the further development of the reaction will occur according to a dynamic scenario: the system can pass through the saddle point (SP), s*, or tunnel through the equilibrium energy barrier on the way to it. On the other hand, if the entire excess kinetic energy at point b is transformed into the C-H bond vibrational energy, the system is able to pass to the final state as a result of tunneling of the H atom from some excited vibrational level of the double-well potential V (Q;r).
In studying reactions (1) – (3), two models of H-atom tunneling were considered [13]. In model 1, it is assumed, in accordance with the GFCP, that the proton transition itself is preceded by symmetrization of the potential V(r; Q). In this case, the reaction is described using a four-stage scheme, including: (1) Bringing the reactants together to a distance Q. (2) Reorganization of the system at a fixed Q. The tunneling coordinate r is used as the structural coordinate, assuming that the motion along r is slow (the coordinate r in this case is assigned the symbol ρ). The position of the activated complex (AC) on the coordinate ρ, ρ* (point c in Figure. 1) corresponds to the symmetrization of the potential V(r; Q). (3) Tunneling of the H-atom in the potential V(r; Q, ρ*) is the motion of the H-atom along the “fast” coordinate rat fixed values of Q and ρ*. Both the second and third stages, occurring, respectively, at one (Q) and two (Q and ρ*) fixed distances, are non-equilibrium. (4) Relaxation of the system to an equilibrium state and separation of the reaction products.
This model describes well the kinetics of the symmetric reaction of methane with the methyl radical (Eq. 4). For the description of the asymmetric reactions of the methyl radical with methanol (Eq. 5 and 6), model 2, in which the processes of H-atom tunneling and system reorganization are considered to occur simultaneously, turned out to be more adequate. In this case, the need to introduce an additional structural coordinate disappears. Accordingly, the reaction can be described using a three-stage scheme, the second (non-equilibrium) stage of which represents the system's movement in an asymmetric potential V(r;Q) (the variable r simultaneously functions as the tunneling and reorganization coordinates). It is assumed that the transition from model 1 to model 2 is associated with a tunneling process slowdown in the asymmetric potential (Sector 4.2).
Regardless of the tunneling model, the equation used to describe the thermal rate constant of the reaction, k(T), is:
k(T) = σ ∫k(Q)dQ = σ ∫ νt(Q,T) exp[-ΔG*(Q)/RT]dQ (7a)
ΔG*(Q) = ΔH*(Q) - T ΔS*(Q) (7b)
ΔH*(Q) = Ea + Δh*(Q) (7c)
ΔS*(Q) = 1000{[Δh*(Q) - Δg*(Q)]/T} (7d)
Δh*(Q) = h*(Q) –h; Δg*(Q) = g*(Q) – g (7e)
To calculate the tunneling frequency νt (Q, T) the following relations are used:
νt(Q,T) = νt 00(Q) + Σi νti (Q,T) (8a)
νti (Q,T) = νtii (Q) exp(-ΔVi0(Q)/RT) (8b)
Here νt 00 and νtii are the H-atom tunneling between zero and i-th levels of the double-well potential V(r;ρ*,Q), respectively, and ΔVi0 is the energy difference between the i-th and zero levels of the potential.
Calculation Details
All calculations were performed using the Gaussian 03 software package [20] at the ener-gy calculation level CCSD (T)/6-311++G**// B3LYP/6-31+G**. Calculations related to the de-termination of tunneling frequencies were performed on the basis of model 2. This is due, as al-ready noted, to the preference of this model for the case of asymmetric reactions, on the one hand, and to the impossibility of performing calculations on the basis of model 1, on the other hand. The reason for this is an extremely high dependence of the system geometry on the struc-tural coordinate ρ, which does not allow fixing the symmetrization point of the tunneling poten-tial.
When optimizing the geometry for the key points of the equilibrium potential V(Q;r): the positions of the left (r01) and right (r02) minima, as well as the barrier top (rb) (schemes 1-3), along with the distance Q, the dihedral angle H1COH7 was also fixed (at a value of 180°). The values of r01 and r02 were taken to be equal to 1.1 and Q - 0.96 A, respectively.
The tunneling frequencies were calculated in the WKB approximation using the Brickman method [21, 22]. The calculations by Eq. 8 are carried out at i≤8. When calculating the thermo-chemical quantities, fixing the above geometric parameters of the system usually leads to the ap-pearance of several (from one to three) imaginary frequencies, which, as assumed, reflects a change in the shape of the vibration potential, namely from a single-well potential to a double-well one. When determining the h* and g* values, the imaginary frequency values were replaced by the corresponding real ones.
The values of the CH3 fragment umbrella vibration frequency, ν(CH3), as well as the C(H)..O bond vibration frequency, ν(CO), in the collision complex (in the left well of the V(r;Q) potential, Scheme 1), were calculated using the GaussView program in the process of calculating thermo-chemical h* and g* parameters.
The procedure for calculating individual parameters (y) included in the integrand of Eq. 7a con-sisted of two stages. In the first stage, the y values were calculated for the studied range of dis-tances Q with a step of 0.1 Å. In the second stage, using the graphically determined dependence y (Q) (in the form of polynomials of various degrees), the y values were obtained with a step of 0.025 Å. The greatest data scatter was observed when calculating the g* values; this required ex-cluding the most deviating g* values when establishing the dependence y(Q), thus limiting the number of variables.
Results and Discussion
Geometrical and Energetic Characteristics of the Reaction; Thermo-Chemical Calculations
The results of calculating the reaction energy, ΔE00, and the barrier height at the saddle point, EbSP, are given in Table 1. At the selected calculation level, the difference between the found parameter values and the corresponding data from high-level calculations [4] is 1.5 – 2.5 kcal.mol-1.
For model 2, the energy Ea is an equilibrium value determined by the energy expenditure in the section ab (Figure. 1), Eaeq:
Ea = Eaeq, (9)
The values of the electron energy Ea and the barrier height Eb for the range of distances Q from 2.5 to 3.0 A are given in Table 2; the position of the barrier top on coordinate r is also given there.
Table 3 demonstrates the changes in the system geometry, the value of the planar angle CH5O (see Schemes) depending on the Q distance and the H-atom position on coordinate r.
The results of the thermo-chemical calculations are given in the Appendix (Tables 1S – 5S).
Tunneling of the H-atom [13, 14]
The description of the H-atom tunneling dynamics in the potential V(r;Q) is associated with the concept of tunneling time, τt [23 ]. In this regard the WKB Buttiker and Landauer equation [24] for the tunneling time of a particle in a symmetric double-well potential V(r) was used as the basis:
τsim = ∫ab{0.5 m/[V(r) – Vi]}0.5 dr (10)
(Vi is the energy of the ith vibrational level and m is the reduced mass of the particle).
Based on the results of kinetic calculations for reactions (4) – (6), it was suggested that in the case of an asymmetric potential barrier, the expression for the tunneling time should be corrected for the asymmetry factor η used in the Brickman WKB model [21, 22]:
η = [1 + (πΔVmin/hν0)2T-2]-0.5 (11)
Where ΔVmin is the difference in the energies of the minima in the potential V(r;Q), ν0 is the zero-point frequency of the H atom vibration in the left well of the potential, and Ttr is the transmis-sion coefficient:
Ttr = ∫ab{0.5 m/[V(r) – Vi]}0.5 dr (12)
According to this assumption, the tunneling time of an H atom in an asymmetric potential V(r;Q) should be described by the equation
τt = τt′/η (13)
Where τt′ is the tunneling time in the potential V(r;Q) without taking into account the value of η.
In the case of the exothermic reaction of methane with the hydroxyl radical, the value of τt was calculated for the zero level in the left well of the V(r;Q) potential and for the distance Q corre-sponding to the maximum of the rate constant k(Q) (Eq. 7a), Qm.
For this reaction (as for reactions (4)–(6)) the main change in the structure of the system is asso-ciated with the transition of the C atom from the sp3 to the sp2 state. Accordingly, the following parameter can be used as a criterion for the dynamic behavior of the system during the tunneling of the H atom:
n = τt /T0.5 (14)
Where T0.5 is the half-period of the umbrella oscillation of the CH3 fragment in the left well of the potential V(r;Q); the T0.5 value defined by the ν(CH3) value (Section 3).
The τt, T0.5 and n values calculation results are given in Table 4. The acquired value of n (> 1) is in agreement with the assumption that in an asymmetric potential the tunneling time of the H atom is relatively (compared T0.5) large; in accordance with model 2, this enables the system structure to adapt to the H-atom motion. The latter implies in this case a significant change in the CH5O angle (Schemes 1 – 3, Table 3).
Kinetics and Mechanism of the Reaction
Calculation of the Rate Constant within the Framework of the Initial Model
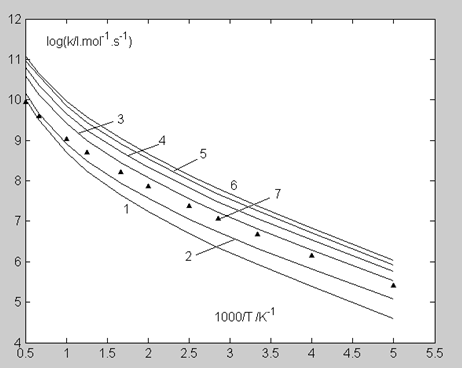
The calculation results of thermal rate constants of the reaction obtained by Eq. 7 are pre-sented in Table 5 (column 2) and Figure. 2 (curve 1). With the exception of the region of high (1500 – 2000 K) temperatures, theoretical values are three to six times lower than the experimental ones (Table 5 (columns 9, 10) and Figure. 2 (curve 7)).
The Promoting Effect
In studies devoted to proton transfer reactions in solution, the heavy atoms vibration of the three-center subsystem A (H)...B is considered as one of the promoting factors [25]. Assuming that this mechanism retains its significance for the reaction in the gas phase (see the next Section), we took as an initial condition that C (H)...O bond five vibrational levels participate in the reaction: the zero and four excited ones. The immediate cause of the reaction acceleration is the decrease in the tunneling distance achieved during the vibration of the C… O bond: the initial tunneling point b (distance Q) shifts to the position b0 (distance Q0) for the zero vibrational level, b1 (distance Q1) for the first level and so on (Figure. 3). The promoting factor, Fj (Q), for a given distance Q and vibrational level j is determined by the relation:
Fj(Q) = νt(Qj)νt(Q)-1 {exp{-[Ej(Q) – E0(Q)]/RT} (15)
Where νt(Q) and νt(Qj) are the tunneling frequencies of the H-atom tunneling at distances Q and Qj, respectively; E0(Q) and Ej(Q) are the energies of the zero and j vibrations at point b.
The rate constant value k (Q) taking into account promotion, kpr(Q), was obtained from the relation
kpr(Q) = k(Q) F(Q) (16)
Where F (Q) is the promotion factor for a given Q:
F (Q) = Σj Fj(Q) (17)
By substituting the kpr(Q) value into Eq. 7a, the resulting value of the thermal rate constant, kpr(T), was determined.
kpr(T) = ∫Q kpr(Q) dQ (18)
The calculations according to Eqs. (15) – (18) were based on the energy profile of the sys-tem along the MEP, presented as a dependence on Q, V(Q), the corresponding dependence for the frequency νt, νt(Q), and the energy E0(Q), determined by the ν(CO) values (Section 3). When calculating the Ej(Q) values, the distance between neighboring vibrational levels, ΔEj,j+1(Q), was considered equal to twice the value of E0(Q): ΔEj,j+1(Q) = 2 E0(Q). The Qj values for the νt(Qj) function (Eq. 15) were found using the inverse V(Q) dependence Q(V) and the energies Ej(Q). In general, the number of vibrational levels in the equation grows up with increasing interval Q - Qmin, where Qmin (= 2.5 A) is the minimum value of Q available for calculation.
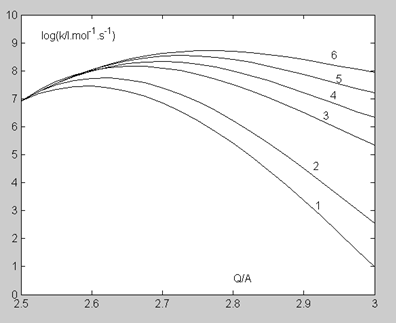
The values of E0 (Q) are given in Table 6. Table 7 provides, as an example, the calculation details of the values of Fj(Q), F(Q) and kpr(Q) at T=500 K and Q=2.8 A . Figure. 4 demonstrates the rate constant k (Q) changes at T=500 K depending on the number of C (H)…O vibrational levels j participating in the reaction.
In the plot for the thermal rate constant (Figure. 2), an increase in the number of vibrational levels involved in the reaction leads to the kinetic curve upward shift; as a result, the entire region of the experimental rate constant is covered. At the same time, the course of the change in the experimental dependence (curve 7) retains a noticeable difference from those predicted by the calculation: at low temperatures, curve 7 is close to curve 3 (the zero and first vibration levels participate in the reaction), and at high temperatures, to curve 1 (there is no promotion). In the next section, the observed behavior of the experimental dependence is considered in connection with the lifetime of the collision complex.
Collision Complex Lifetime (τlf)
The Bunker equation [26], obtained on the basis of real gases physical properties, predicts a decrease in the τlf value with temperature:
τlf = 1.50 σ μ1/2 ε1/6 (2kBT)-2/3 (19)
Here σ is the collision diameter, ε is the well depth for the Lennard-Jones potential, μ is the reduced mass of the complex, and kB is the Boltzmann constant. The values of the σ and ε parame-ters for the reaction collision complex were obtained on the basis of tabulated values for the reac-tants [see, for example, [25]): σ as the arithmetic mean, and ε as the geometric mean (Table 8). As the calculation shows, in the temperature range from 200 to 2000 K, the τlf value changes from 4.5 to 1.0 10 -13 sec (Table 9). For comparison, the oscillation period of the C (H)...O bond, Tvb, at Q = 2.6 A (corresponding to the maximum of the k (Q) value at all temperatures) is 1.3 10-13 sec. accordingly, with increasing temperature the ratio p= τlf/Tvb changes from 3.5 to less than 1 (Table 9). These changes are in obvious agreement with the transition from the promotion to its complete absence described in the previous section.
The variable parameter f (Table 9) shows the change in lifetime relative to its value at 200 K:
f = τlf/τlf(200 К) (20)
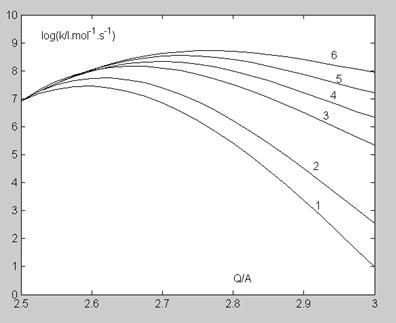
The rate constant k1 adjusted for this correction, k1′ (Eq. 21), shows a close agreement with the experiment for the entire studied temperature range (Figure. 5, Table 5).
k1′ = k1 f (21)
The direct influence of the collision complex lifetime on the reaction rate observed in this case apparently reflects a change in the population of the C (H)…O bond vibrational levels.
Thus, the performed kinetic analysis allows us to identify several dynamic characteristics of the reaction:
(1) The process of H-atom tunneling at a fixed C...O distance occurs with simultaneous re-organization of the system.
(2) The reaction process occurs with the formation of a collision complex;
(3) The motion of the system along the MEP occurs both due to the translational energy of the reagents and due to vibrations of the C (H)...O bond;
(4) Two vibrational levels of the C (H)...O bond mainly participate in the reaction: zero and first ones;
(5) When temperature increases, the promoting effect of vibration is limited by of the collision complex lifetime.
Taking into account the insufficient accuracy of some calculations (Sections 3 and 4.1), it appears that the observed agreement between theory and experiment for the thermal rate constants (Figure. 5 and Table 5) should be partly attributed to the mutual compensation of calculation errors. It seems, however, that the obtained results are significant and in total confirm the realism of the non-equilibrium approach to studying the gas phase H-atom transfer reactions.
References
- D L Baulch, et al. (1986) Evaluated kinetic data for high-temperature reactions. Volume 5. Part 1. Homogeneous gas phase reactions of the hydroxyl radical with alkanes, J. Phys. Chem. Ref. Data 15: 465-592.
- DL Baulch, et al. (2005) Evaluated kinetic data for combustion modeling: Supplement II, J. Phys. Chem. Ref. Data, 34: 757 – 1397.
- BJ Lynch, PL Fast, M Harris, DG Truhlar (2000) Adiabatic connection for kinetics, J. Phys. Chem. 104: 4811-481.
- J Espinosa-Garcia, JC Corchado, (2015) QCT dynamics study of the reaction of hydroxyl radical and methane using a new ab initio fitted full-dimensional analytical potential energy surface, Theor. Chem. Acc. 134: 6.
- JLi, H Guo, (2015) Communication: An accurate full 15 dimensional permutationally invariant potential energy surface for the OH + CH4→ H2O + CH3 reaction, J. Chem. Phys. 143: 221103
- YV Suleimanov, J Espinosa-Garcia (2016) Recrossing and tunneling in the kinetics study of the OH + CH4→ H2O + CH3 reaction, J. Phys. Chem. B 120: 1418-28.
- Li J, Guo H (2018) Thermal rate coefficients and kinetic isotope effects for the reaction OH + CH4→ H2O + CH3 on an ab initio-based Potential Energy Surface, J. Phys. Chem. A 122: 2645-52
- Allen J W, et al. (2013) Communication: Full dimensional quantum rate coefficients and kinetic iso-tope effects from ring polymer dynamics for a seven-atom reaction OH + CH4→ CH3 + H2O, J. Chem. Phys. 138: 221103.
- H. Song, et al. (2014) Effects of reactant rotation on the dynamics of the OH + CH4→ H2O + CH3 reaction: A six-dimensional study, J. Chem. Phys. 140: 084307.
- J Espinosa-Garcia, J C Corchado (2016) Product translational and vibrational distributions for the OH/OD + CH4/CD4 reactions from quasiclassical trajectory calculations. Comparison and ex-periment, J. Phys. Chem. B 120: 1446-53.
- IA Romanskii (2022) Study of gas-phase reactions within the modified Marcus model. IV. Arrhe-nius equation for the reaction CH4 + CH3 → CH3 + CH4, Reaction Kinetics, Mechanisms and Catalysis 135: 2401-23.
- IA Romanskii (2023) Study of gas-phase reactions within the modified Marcus model. V. Ar-rhenius equation for the reaction CH3OH + CH3 → CH2OH + CH4, Univers J. Catal. Sci. 1: 110-130.
- I Romanskii (2024) Investigation of methyl radical with methane and methanol reactions in the framework of a non-equilibrium approach; dynamic aspect, Comp. and Theor. Chem. 1239: 114725.
- I Romanskii (2025) Corrigendum to”Investigation of methyl radical with methane and methanol reactions in the framework of a non-equilibrium approach; dynamic aspect”, Comp. and Theor. Chem.
- RA Marcus (1957) on the theory of oxidation - reduction reactions involving electron transfer. II. Applications to data on the rates of isotopic exchange reactions, J. Chem. Phys. 26: 867–71.
- RA Marcus (1968) Theoretical relations among rate constants, barriers and Broensted slopes of chemical reactions, J. Phys. Chem. 72: 891-99.
- VG Levich, RR Dogonadze, ED German, et al. (1970) Theory of homogeneous reactions involving proton ransfer, Electrochimica Acta 15: 353–67.
- ED German, RR Dogonadze (1977) Quantum-mechanical theory of the kinetics of proton transfer reactions, Supplement to R. P. Bell, The proton in chemistry, Mir, Moscow: 350–76.
- RR Dogonadze, AM Kuznetsov (1974) Theory of the elementary act of charge transfer re-actions in polar solvents, Mendeleev Chem. J. 19: 242–50.
- MJ Frisch, GW Trucks, HB Schlegel, GE Scuceria, et al. (2004) Gaussian 03. (Revision C.02), Gaussian Inc., Wallingford CT.
- J Brickmann, in: P Schuster, G Zandel, C Sandorfy (1976) Proton Motions in Hydro-gen Bond – Recent Developments in Theory and Experiments. North-Holland Publishing Co., Amsterdam: 217–43.
- MM Szczesniak, S Scheiner (1985) Effects of externial ions on the dynamics of proton transfer across a hydrogen bond, J. Phys. Chem. 89: 1835–40.
- K Maji, CK Mondal, SP Bhattacharyya (2007) Tunneling time and tunneling dynamics, International Reviews in Physical Chemistry 26: 647-70.
- M Buttiker, R Landauer (1982) Traversal Time for Tunneling, Phys. Rev. Lett. 49: 1739-42.
- MV Bazilevsky, MV Vener, Usp Khim. 72 [Russ. Chem. Rev. 72 (2003) (Engl. Transl.)].
- DL Bunker (1960) Mechanics of atomic recombination reactions, J. Chem. Phys. 32: 1001–5.
- AW Jasper, JA Miller (2014) Lennard-Jones parameters for combustion and chemical kinetics modeling from full-dimensional intermolecular potentials, Combust. Flame 161: 101-10.
Article Information
Research Article
Received Date: January 05, 2026
Accepted Date: January 20, 2026
Published Date: January 23, 2026
Study of Hydrogen Transfer Reactions within the Framework of the Non-Equilibrium Approach; CH4 + OH → CH3 + H2O Reaction
Volume 1 | Issue 1
Citation
Igor Romanskii (2026) Study of Hydrogen Transfer Reactions within the Framework of the Non-Equilibrium Ap-Proach; CH4 + OH → CH3 + H2O Reaction. World J Immunol and Resp Med 1:101
Copyright
©2026 Igor Romanskii. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.

doi: jcrc.2026.1.101