

Review Article
Volume-1 Issue-1, 2021
Chromosome Anomalies in Human Oocytes and Embryos: From Practice of Preimplantation Genetic Testing for Aneuploidy
Received Date: January 19, 2022
Accepted Date: February 19, 2022
Published Date: February 22, 2022
Journal Information
Abstract
Chromosomal abnormalities originate predominantly from female meiosis, so the direct analysis of the outcome of the first and second meiotic divisions allows performing preimplantation genetic testing for aneuploidy (PGT-A). This approach to PGT-A revealed novel information on the prevalence of meiotic errors and significantly different chromosomal profile from those described in previous meiotic studies, allowing a high accuracy of the oocyte genotype prediction in pre-selection of embryos resulting from aneuploidy-free oocytes. Although, this is no longer a standard PGT-A practice, its utilization is still the only choice in the population settings, where embryo manipulation is not allowed. It also provides a unique information on the origin and mechanism of human chromosomal abnormalities detected in the current PGT-A based on NGS and blastocyst biopsy. It is particularly essential in distinguishing meiosis or mitosis origin of the resulting embryo mosaicism in preselection and prioritization of transfer of such embryos, to ensure the highest potential for pregnancy and birth of a healthy offspring.
Key words
Chromosomal aneuploidy; Meiosis I; Meiosis II; Polar Body Analysis; Preimplantation Genetic Testing for aneuploidy (PGT-A); Mosaicism
Embryos Tested |
Total 20,269 |
||
Euploid |
Total |
8,795 |
43.4% |
Aneuploid |
Monosomy |
2,793 |
27.5% |
Trisomy |
3,078 |
30.31% |
|
Complex |
3,412 |
33.6% |
|
Full Segmental |
853 |
8.4% |
|
Total |
10,136 |
50.10% |
|
Mosaic |
Whole chromosome |
1051 |
78.56% |
Segmental |
123 |
9.23% |
|
Complex |
164 |
12.22% |
|
Total |
1338 |
6.6% |
|
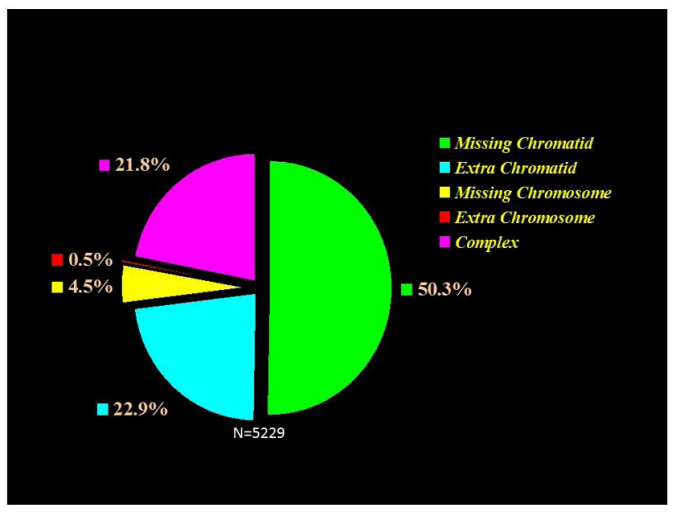 |
| Figure 1: Meiosis I errors, demonstrating predominance of chromatid abnormalities, with significantly excess of nullisomies over disomies |
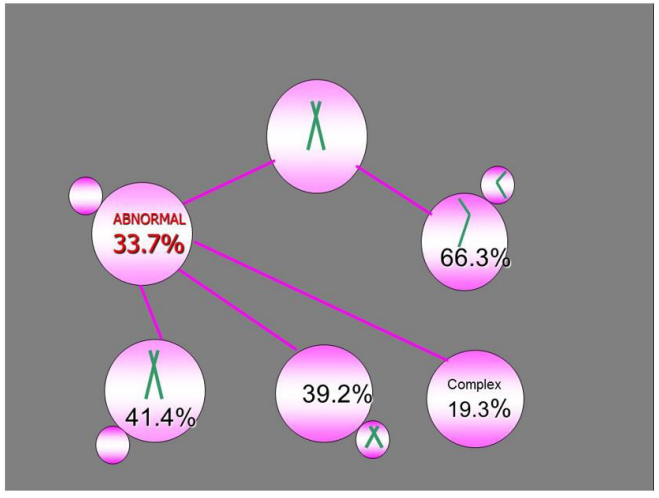 |
| Figure 2:Meiosis II errors with comparable ration of missing and extra chromatids |
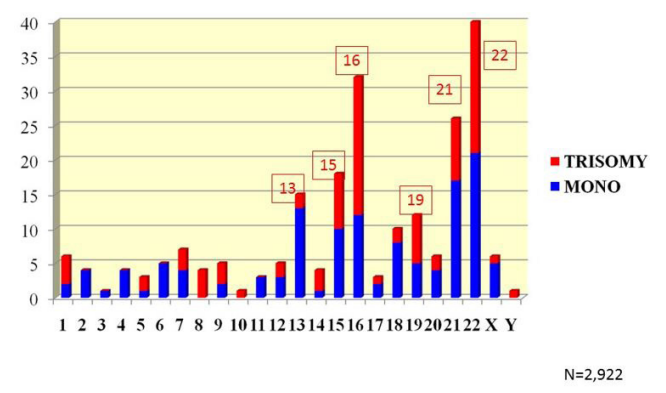 |
| Figure 3:Chromosome specific prevalence of Monosomy and Trisomy Detected in Day-5 Embryos |
Introduction
It is well known that chromosomal abnormalities originate predominantly from female meiosis, and derive mainly from meiosis I [1-3]. It is also agreed that aneuploidy prevalence is maternal age depended due to age-related reduction of meiotic recombination, resulting in premature separation of bivalents and chromosomal non-disjunction. Meiosis II errors was also thought to originate from meiosis I, but on the contrary, as a result of the increased meiotic recombination rate, leading to a separation failure of bivalents [4]. Thus, the direct analysis of the outcome of the first and second meiotic divisions, using the first and second polar body (PB1; PB2) was used for preimplantation genetic testing for aneuploidy (PGT-A) [5-6]. This was based on the concept that PB1, extruded following maturation of oocytes, represents a by-product of meiosis I, while PB2 is a by-product of meiosis II, naturally extruded following the exposure of oocytes to sperm or ICSI. As will be described below, prevalence and types of chromosomal errors detected by this approach are different from what was described in the earlier traditional studies of meiotic chromosomes in metaphase II oocytes, according to which chromosomal anomalies in oocytes originate mainly from the errors of whole bivalents as result of chromosomal non-disjunction [7]. As will be described below, on the contrary, direct testing of meiosis process outcome has revealed not only a higher prevalence of meiotic errors, but also a significantly different chromosomal profile, allowing a high accuracy of the oocyte genotype prediction for pre-selection of embryos resulting from aneuploidy-free oocytes [8-11].
As PBs are extruded as a natural process, their removal requires the least invasive intervention, in pre-selection of zygotes with a higher developmental potential. This traditionally was based on pre-selection of embryos by morphological parameters which are of a limited value for this purpose. Although PB-based PGT-A is no longer a standard of practice either, its utilization is still the only choice in the population settings, where embryo manipulation is not allowed. This also provides the unique information on the origin and mechanism of human chromosomal abnormalities, which is described below. In addition, with the improvement of resolution in detection of copy number variation in the current PGT-A based on the next generation sequencing (NGS) coupled with blastocyst biopsy procedure, a sizeable group of detected embryo mosaicism presents the problem in decision making on the transfer of such embryos, requiring the information on their origin provided by PB analysis, to insure the expected reproductive outcome [12].
Prevalence and Types of Chromosomal Abnormalities in Human Oocytes
Based on our data of 20,000 oocytes [8-11], tested in process of PGT-A, approximately half of these oocytes were chromosomally abnormal, originating either from both meiosis I and meiosis II, either from meiosis I, or from meiosis II only, in approximately equal proportions. The frequency of aneuploidies of meiosis origin increases with increasing of maternal age, from up to 20% in patients of 35 years of age, to over 40% in patients older than 40, which is in agreement with other studies [13-14]. It is not clear to what extent the reported meiotic aneuploidies are related to IVF treatments involving aggressive hormonal stimulation, as the actual prevalence may be much lower in donated oocytes from young fertile women [15].
In contrast to the well-established concept of female meiosis I origin of the majority of aneuploidies, PB-based PGT-A showed a comparable proportions of detectable aneuploidies originating from meiosis I (31,0%) and meiosis II (33.7%), [8-11]. It is of interest, that a one-third of the chromosomally abnormal oocytes originate from sequential meiosis I and meiosis II errors, suggesting that these meiosis II errors may be related to the preceding meiosis I errors. This is in agreement with the concept of a possible relationship of meiosis II errors with the increased meiotic recombination rate mentioned above [4]. However, another half of meiosis II errors were independent from meiosis I, with all types of errors showing the maternal age dependence. This is of clinical significance, as clearly the genotype of the resulting zygote cannot be predicted without testing of the outcomes of both meiotic divisions. Of course, even testing of meiosis I errors alone could reduce aneuploidy rates in the resulting embryos by two-thirds, but over one-third of these oocytes will be still abnormal following the second meiotic division. Thus, testing for only MI errors may still improve the implantation and pregnancy rates in poor prognosis IVF patients, by applying ICSI selectively only to the oocytes with aneuploidyfree PB1, despite that only half of the abnormalities deriving from the second meiotic division may be detected following MI. The spectrum of types of errors resulting from MI showed a two times higher frequency of nullisomies over disomies (Figure 1), in contrast to a comparable nullisomy/ disomy ratio after MII (Figure 2). Thus, missing chromosome materials in the extruded PB1 are more frequent than extra chromosomes, in agreement with a higher frequency of trisomies over monosomies in spontaneous abortions. Such a predominance of meiosis I originating nullisomies may be due to a possible meiosis I check point mechanism, preventing extra chromosome material extrusion during oocyte maturation, unless it is attributable to a technical error. This, however, may be excluded, as all the detected types of errors show a strong maternal age dependence [8-11] that may suggest a biological nature of this phenomenon caused by the overall disturbances of the meiosis process with age.
As mentioned, the majority of aneuploidies originating from meiosis I are represented by chromatid errors (Figure 1), in contrast to the expected chromosomal nondisjunction, as suggested by previous traditional studies. However, chromosomal errors are still observed in sizable proportion (6.3%) of oocytes, thus the abnormalities in MII oocytes are not solely of chromatid origin. Although both chromatid and chromosomal errors are involved in producing metaphase II oocyte abnormalities, the chromatid/chromosome error ratio is as high as 10:1. Clearly, both of these meiosis I errors lead to aneuploidy in the resulting embryos, with no data available on their possible differences on the pre- and post-implantation development. On the contrast to meiosis I errors, there is no difference in the frequency of missing or extra chromatid errors following meiosis II (Figure 2).
While at least 95% of aneuploidies originate from maternal meiosis, the remaining aneuploidies derive from paternal meiosis and/or mitotic errors. To cover the whole spectrum, as mentioned, the standard PGT-A methodology is currently based on the next generation sequencing (NGS) following blastocyst biopsy. This is also more practical, because not all oocytes can reach the embryo transfer stage, with also selection against aneuploid embryos that may not survive to blastocyst. In fact, it is of note that there is an inconsistency between the expected and observed frequency of some types of aneuploidies in embryos as predicted by meiosis testing. While the predicted lower rate of monosomies is in agreement with the lack of autosomal monosomies in spontaneous abortions, due to incompatibility with post-implantation development, the predicted embryo trisomy predominance is not in accordance to the observed trisomy prevalence in the resulting embryos, being comparable to monosomy rate (Table 1). It is of interest that the latter did not show the maternal age dependence, suggesting a possible artefactual nature of monosomies detected by PGT-A [11].
This discordance may be explained by the possibility that the majority of monosomies detected in embryos are deriving from mitotic errors. In fact, a significant proportion of the cleavage-stage monosomies were shown to be euploid after their reanalysis [16]. The fact that almost no embryo monosomies are detected after implantation, may be due to their elimination before implantation. However, the majority of pre-zygotically derived monosomies, as well as at least some of postzygotic origin, may still survive until the blastocyst stage, therefore leading to implantation failure or pregnancy loss. A follow up of progression and survival of over two thousand fertilized oocytes with different types of chromosome abnormalities did not detect the initial variety of chromosome abnormalities later in development, although well tolerated until activation of the embryonic genome [17]. However, many aneuploid embryos still successfully reach the blastocyst stage, even if some chromosome errors present during preimplantation development are not detected in clinically recognized pregnancy.
Of special interest is the group of complex aneuploidies, detected in one-fifth of abnormalities following meiosis I and II (Figures 1 and 2). These abnormalities involve the errors in both MI and MII of the same chromosome that may result in balanced (normal) embryos, representing the phenomenon of aneuploidy rescue, similar to the well-known trisomy rescue mechanism. Although the resulting chromosome set of the oocytes after complementary (reciprocal) errors in meiosis I and meiosis II may be considered balanced, the fate of these embryos is not clear and may result into abnormal (mosaic) status, uniparental disomy and imprinting disorders.
Overall, the high prevalence of complex errors may indicate generalized disturbances in the meiosis process due to the age-related effect on the recombination frequency, spindle formation errors that are also reported to increase with age, loss of chromosome cohesion and mitochondrial and organelle dysfunction [18-24]. However, the actual mechanism underlying correlation with maternal age is still not understood. The data show a strong correlation between female ageing and premature resolution of centromeric cohesion, which is characterised by depletion of the meiosis-specific alpha-kleisin subunit Rec8 from the oocyte chromosomes. Together with the involvement of other meiosis-specific proteins, this event may explain the prevalence of premature loss of centromeric cohesin in oocytes of older females. Analysis of the timing and mechanisms of cohesin depletion during female ageing, show that cohesin is gradually depleted from the oocyte chromosomes during the prolonged arrest at prophase of meiosis I, long before oocytes are recruited for growth.
Chromosome-Specific Meiotic Error Origin and Its Impact on Embryo Viability
Analysis of chromosome-specific patterns has shown that chromosomes 15, 16, 21 and 22, are much more frequently involved in female meiosis errors than other 20 chromosomes, representing over one third of all oocyte aneuploidies. This is followed by chromosomes 19 and 20, with errors of other 18 chromosomes being of lower frequency [24-26]. These data are in agreement with data obtained in PGT-A performed at the blastocyst stage (Figure 3). It was also previously demonstrated that despite differences in chromosome-specific aneuploidy rates, the maternal age dependence was observed for each of these chromosome errors, almost doubling between the age 35 and 43 years, again suggesting overall disturbance of the meiosis process with advanced reproductive age [11].
Chromosome-specific origin of errors was also not similar: chromosome 16 and 22 errors originated more frequently in meiosis II (44.4% and 41.5% meiosis II errors, vs. 32.0% and 34.3% meiosis I errors, respectively). Chromosome 13, 18, and 21 errors were more frequently from meiosis I (40.1%, 48.3%, and 41.4% in meiosis I vs. 36.3%, 34.6%, and 36.7% in meiosis II, respectively). It is of note that the proportion of oocytes with errors of both meiosis I and meiosis II origin, were not significantly different for errors of different chromosomes, except for chromosome 18 errors [8-11].
These data are opposite to the chromosome specific meiosis origin observed in spontaneous abortions and live-born children [2] and may indicate poor viability of embryos resulting from the oocytes with the chromosome 16 and 22 errors of the second meiotic division, possibly incompatible with implantation and post-implantation development. At present, there is no explanation for the possible biological differences of aneuploidies depending on the meiotic origin, except for loss of heterozygosity or higher homozygosity of the embryos originating from meiosis II errors for the genes located on these chromosomes. This may lead to imprinting of paternal or maternal genes on chromosomes 16 or 22, although there is no sufficient data on the established imprinting genes in these chromosomes.
Whatever may be the explanation for the above phenomena, these data provide evidence for a possible viability differences depending upon not only the chromosome involved but also the meiotic origin of the error.
Chromosomal Anomalies in Embryos in Relation to Meiosis Errors
As shown above, approximately half of meiosis II errors are observed in the oocytes with prior errors in meiosis I, and as a result of such sequential errors, one-third of the resulting zygotes may have been considered normal (euplolid), provided that the preceding errors in meiosis I and meiosis II have no effect on the further developments of the corresponding embryos. Based on this observation, it could have been postulated that a pooled testing of both polar bodies could be acceptable, as it will show the normal results in the cases PB1 and PB2 reciprocal errors, or show an abnormality if the errors are not balanced or occur only in the first or second meiotic divisions. The follow-up testing of these embryos with reciprocal MI and MII errors showed that only 18% embryos, deriving from such apparently balanced zygotes, were euploid for all the chromosomes analyzed [8-11].
As the average reproductive age of the patients from whom the oocytes were obtained was approximately 38.5 years, the observed genomic instability in mitotic divisions of the apparently balanced zygotes following meiosis II rescue may be age related. The above mentioned advanced maternal age effects may also apply to increasing errors in the mitotic machinery of dividing cells. It has been suggested that deviations in the cytoplasmic organization, such as mitochondrial distribution and maternal genetic variants spanning the centrosomal regulator PLK4, may reduce meiotic competence of oocytes and predispose the embryos to common cleavage abnormalities [27- 30]. The relationship between these cytoplasmic changes and the nuclear organization during maturation and fertilization of oocytes may determine an abnormal development and mitotic errors, as suggested in prospective analysis of pronuclear zygote morphology in relation to chromosomal abnormalities detected in PGT for poor prognosis IVF patients [31,32].
Because there is usually no information about the initial chromosomal set of zygotes from which mosaic embryos originated, the nature of mosaicism in preimplantation embryos is not known. There are, however, indirect observations suggesting that the observed mosaicism may be of different nature. Some mosaic types increase with maternal age [33], and therefore probably stemming from female meiosis errors. Others are possibly attributable to immaturity of centrosome structures in sperm, expected to be active from the first mitotic divisions of zygote as suggested for the cases involving patients requiring microsurgical testicular sperm extraction (TESE) [34].
In a recent PGDIS position statement on mosaicism mentioned, which was based on the analysis of the follow up of one thousand transferred mosaic embryos, none appeared to lead to live births with detected no neonatal abnormalities [12,35]. However, the implantation ability of a mosaic embryo depended not on the specific chromosome involved, but the proportion of euploid cells present. If the vast majority of cells are chromosomally normal, these will dominate over aneuploid cells, thusfacilitating ability to implant and lead to a chromosomally normal live birth. The converse would apply if the vast majority of cells are aneuploid. The latter mosaic embryos either fail to implant or are destined to be lost early in pregnancy. Thus, a low-level mosaic embryos have a significant potential to reach term, and could be considered for transfer in the absence of euploid embryos. It may be suggested that the majority of low-level mosaics may have actually been a false positive, as has been demonstrated in the extended in vitro culture of mosaic embryos to the day 12, 71% of which appeared to be normal both in trophectoderm and inner cell mass [36]. On the contrary, the other study revealed a meiosis error as an origin of mosaicism, and therefore should be avoided from transfer, to prevent the poor transfer outcome of this mosaic embryo [37]. Thus, the development of reliable additional testing will allow pre-selection and prioritization of transfer of mosaic embryos to ensure the highest potential in achieving pregnancy and birth of a healthy offspring.
In conclusion, presented data on the types of chromosomal aneuploidies and the prevalence of each chromosome-specific error in oocytes and embryos further suggest that most chromosomal aneuploidies in embryos originate from female meiosis, predisposing to further sequential post-zygotic errors, which may explain the high rate of chromosomal instability in preimplantation embryos. This may also call for requirements of detection of the origin of aneuploidies, having important value in pre-selection and prioritization of single embryo transfer with highest potential to result in healthy pregnancy.
References
- Sherman SL, Peterson MB, Freeman SB (1994) Nondisjunction of chromosome 21 in maternal meiosis I: evidence for a maternal age-dependent mechanism involving reduced recombination. Hum Mol Genet 3: 1529-35.
- Hassold T, Merril M, Adkins K, Freemen S, Sherman S (1995) Recombination and maternal age-dependent nondisjunction: molecular studies of trisomy 16. Am J Hum Genet 57: 867-74.
- Peterson MB, Mikkelsen M (2000) Nondisjunction in trisomy 21: origin and mechanisms. Cytogenet Cell Genet. 91: 199–203.
- Lamb NE, Freeman S, Savage-Austin A (1996) Susceptible chiasmate configurations of chromosome 21 predispose to nondisjunction in both maternal meiosis I, and meiosis II. Nat Genet 14: 400–5.
- Verlinsky Y, Kuliev A (1993) Preimplantation diagnosis of genetic disorders: a new technique for assisted reproduction. New York: Wiley Liss.
- Verlinsky Y, Kuliev A (2000) Atlas of preimplantation genetic diagnosis. New York, London: Parthenon.
- Pellestor F, Andreo B, Armal F, Humeau C, Demaille J (2002) Mechanisms of non-disjunction in human female meiosis: the co-existence of two modes of malsegregation evidenced by the karyotyping of 1397 in-vitro unfertilized oocytes. Hum Reprod 17: 2134–45
- Kuliev A, Cieslak J, Verlinsky Y (2005) Frequency and distribution of chromosomal abnormalities in human oocytes. Cytogenet Genome Res 111: 193-8.
- Kuliev A, Zlatopolsky Z, Kirillova I, Spivakova J, CieslakJanzen G (2011) Meiosis errors in over 20,000 oocytes studied in the practice of preimplantation aneuploidy testing. Reprod Biomed Online 22: 2–8.
- Kuliev A, Rechitsky S, Verlinsky O (2020) Atlas of preimplantation genetic diagnosis. 3rd Edition; 20CRC Press.
- Kuliev A, Rechitsky S, Simpson JL Practical Preimplantation Genetic Testing. Springer, Nature 2020
- D Leigh, DS Cram, S Rechitsky, A Handyside, D Wells, et al. (2022) PGDIS Position Statement on the Transfer of Mosaic Embryos Reprod Biomed Online (in press).
- Magli MC, Gianaroli L, Crippa A, Grugnetti C, Ruberti A, et al. (2009) Causes of aneuploidy – polar body based PGD. Reprod Biomed Online 18: S3.
- Gianaroli L, Magli MC, Lappi M, Capoti A, Robles F, et al. (2009) Preconception diagnosis. Reprod Biomed Online 18: S5.
- Fragouli E, Escalona E, Guttieres Mateo C (2009) Comparative genomic hybridization of oocytes and first polar bodies from young donors. Reprod Biomed Online 19: 228–37.
- Uher P, Baborova P, Kralickova M, Zech MH, Verlinsky Y, et al. (2009) Non-informative results and monosomies in PGD: the importance of a third round of re-hybridization. Reprod Biomed Online 18: 530-46.
- Fragouli E, Alfarawati S, Spath K (2013) The origin and impact of embryonic aneuploidy. Hum Genet 132: 1001-13.
- Hassold T, Hall H, Hunt P (2007) The origin of human aneuploidy: where we have been, where we are going. Hum Mol Genet 16: 203–208.
- Lamb NE, Feingold E, Savage-Austin A (1997) Characterization of susceptible chiasma configurations that increase the risk for maternal nondisjunction of chromosome 21. Hum Mol Genet 6: 1391-401.
- Battaglia DE, Goodwin P, Klein NA, Soules MR (1996) Influence of maternal age on meiotic spindle assembly in oocytes from naturally cycling women. Hum Reprod 11: 2217–22.
- Eichenlaub-Ritter U, Vogt E, Yiu H, Gosden R (2002) Spindles, mitochondria and redox potential in ageing oocytes. Reprod Biomed Online 5: 117–124.
- Chatzimeletiou K, Morrison EE, Prapas N, Prapas Y, Handyside AH (2005) Spindle abnormalities in normally developing and arrested human preimplantation embryos in vitro identified by confocal laser scanning microscopy. Hum Reprod 2005; 20: 672–82.
- Angel E, Antonarakis SE (2002) Genomic imprinting and uniparental disomy in medicine: clinical and molecular aspects. New York: Willey Liss; 2002
- Herbert M (2019) How and when do oocyte chromosomes fall apart during female ageing? Reprod Biomed Online, 39: e8-e9
- Handyside A, Montag M, Magli C (2012) Multiple meiotic errors caused by predivision of chromatids in women of advanced maternal age undergoing in vitro fertilization. European J Human Genetics 20: 742-7
- Handyside A, Montag M, Magli C (2012) Multiple meiotic errors caused by predivision of chromatids in women of advanced maternal age undergoing in vitro fertilization. European J Human Genetics 20: 742-7
- Lamb NE, Feingold E, Savage-Austin A (1997) Characterization of susceptible chiasma configurations that increase the risk for maternal nondisjunction of chromosome 21. Hum Mol Genet 6: 1391–401.
- Van Blercom J, Davis P, Alexander S (2000) Differential mitochondrial distribution in human pronuclear embryos leads to disproportionate inheritance between blastomeres: relationship to microtubular organization, ATP content and competence. Hum Reprod 15: 2621–33.
- McCoy RC, Newnham LJ, Ottolini CS (2018) Tripolarchromosome segregation drives the association between maternal genotype at variants spanning PLK4 and aneuploidy in human preimplantation embryos. Hum Mol Genet 27: 2573-85
- Kim NH, Chung HM, Cha KY, Chung KS (1998) Microtubule and microfilament organization in maturing human oocytes. Hum Reprod 13: 2217–22.
- Kahraman S, Kumpete Y, Sertyel S (2002) Pronuclear scoring and chromosomal status of embryos in severe male infertility. Hum Reprod 17: 3193–200.
- Gianaroli L, Magli MC, Ferraretti AP (2003) Pronuclear morphology and chromosomal abnormalities as scoring criteria for embryo selection. Fertil Steril 80: 837–44.
- Munne S, Sandalinas M, Escudero T (2002) Some mosaic types increase with maternal age. Reprod Biomed Online 4: 223–32.
- Silber S, Sadowy S, Lehahan K, Kilani Z, Gianaroli L, et al. (2002) High rate of chromosome mosaicism but not aneuploidy in embryos from karyotypically normal men requiring TESE. Reprod Biomed Online 4: 20.
- Viotti M, Victor AR, Barnes FL, Zouves CG, Besser AG, et al. (2021) Using outcome data from one thousand mosaic embryo transfers to formulate an embryo ranking system for clinical use. Fertil. Steril 115: 1212-24.
- Popovich M, Dhaemens L, Thelman J (2019) Expanding In vitro culture of human embryos demonstrated the complex nature of diagnosis chromosomal mosaicism from single trophectoderm biopsy. Hum Reprod, 2019: 1-12.
- A Handyside, A McCollin, M Summers, CS Ottolini (2021) Copy number analysis of meiotic and postzygotic mitotic aneuploidies in throphectoderm cell biopsied at the blastocyst stage and arrested embryos. Prenatal Diagnosis, 41: 525-53
Artcle Information
Review Article
Received Date: January 19, 2022
Accepted Date: February 19, 2022
Published Date: February 22, 2022
Journal of Genetic Diseases and Therapeutics
Volume 1 | Issue 1
Citation
Anver Kuliev (2022) Chromosome Anomalies in Human Oocytes and Embryos: From Practice of Preimplantation Genetic Testing for Aneuploidy. J Genet Dis Ther 1: 1-9
Copyright
©2022 Anver Kuliev. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.

doi: jgdt.2022.1.101