

Review Article
Volume-1 Issue-1, 2021
Case Report: Array CGH Analysis Shows Pathogenic Homozygote Deletion of FANCA Gene Detected in a New Born with Multiple Congenital Abnormalities
Received Date: June 13, 2021
Accepted Date: July 15, 2021
Published Date: July 17, 2021
Journal Information
Abstract
Fanconi anemia (FA, Omim#227650) is a rare autosomal recessive or X-Linked chromosomal instability disorder caused by mutation in one of seven known genes (FANCA, C, D2, E, F, G and BRCA2). Affected individuals have highly variable clinical manifestations characterized by congenital abnormalities, progressive bone marrow failure with an increasing susceptibility to hematological malignancies and solid tumors of the head and neck. In this study, we present a case of dead baby with major CNS anomalies and renal anomalies with homozygote deletion into FANCA (Omim#607139) gene diagnosed by chromosomal Microarray analysis (Agilent Technology).
Array CGH result of both amniotic fluid and peripheral blood from the baby showed a homozygote deletion of 45 Kb on the long arm of chromosome 16 at q24.3 (arr[GR CH37]16q24.3(89851577_89897040)x0) into FANCA gene including the first exons of the gene.
Deletion of the FANCA gene have been extensively reported from exon 1 to exon 18, however, the homozygote deletion is never reported before. The deletion result from recombination between very distant Alu repeats and involve 2 adjacent genes SPIRE2 and TCF25, which heterozygous deletion would not cause dominant inherited disease, however the homozygote deletion give more severe phenotype as our case, the patient died at the age of one year and 4 months. In addition, our CNV result is not detected by WES.
Key words
Fanconi Anemia; FANCA Gene Deletion; Multiple Congenital Malformation; AGH; WES; Chromosomal Analysis
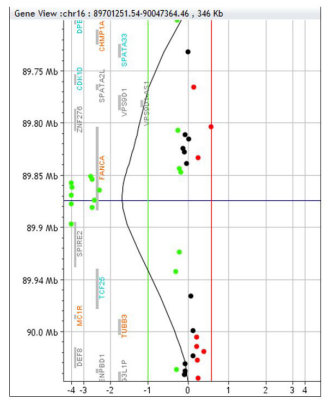 |
| Figure 1: Result of 180 K Agilent micro-array analysis of patient DNA showing 45kb homzygote deletion of FANCA gene including 2 adjacent genes SPIRE2 and TCF25 |
 |
| Figure 2:Fluorescent in-situ hybridization (FISH) result shows normal results (a) 13 spectrum green & 21 spectrum orange; (b) 18 spectrum aqua, X spectrum green & Y spectrum orange |
 |
| Figure 3:Chromosome 16 |
Introduction
Fanconi anemia FA) is a rare inherited autosomal recessive disorder, resulting from mutations in one of at least 16 known FA genes. Mutations in these genes are the most common feature causing Fanconi anemia. This disease is characterized by congenital malformations, progressive bone marrow failure, aplastic anemia in childhood and other cancers. It affects 1 in 130,000 births. The most significant characteristic of FA cells is particularly responsible to a certain type of DNA damage known as Interstrand cross-links. Generally cells from Fanconi anaemia patients show a markedly higher frequency of spontaneous chromosomal breakage and hypersensitivity to the clastogenic effect of DNA cross-linking agents such as diepoxybutane (DEB) and mitomycin-C (MMC) when compared to normal cells. The primary diagnostic test for Fanconi anemia is based on the increased chromosomal breakage seen in afflicted cells after exposure to these agents – the DEB/MMC stress test.
Fanconi Anemia Complementation group A, also known as FAA, FACA and FANCA, is a protein which in humans is encoded by the FANCA gene and mutation in this gene is responsible for 60% of Fanconi Anemia disease. Mutations involving the FANCA gene are associated with many somatic and congenital defects, primarily involving phenotypic variations of Fanconi anemia, aplastic anemia, and forms of cancer such as squamous cell carcinoma and acute myeloid leukemia.
We here present a case of an amniotic fluid with 33 weeks gestational age, single pregnancy and multiple congenital anomalies for the baby. The amniotic fluid was processed for both FISH aneuploidy and microarray. The FISH for aneuploidy was normal and Array CGH result showed a homozygote deletion of 45 Kb on the long arm of chromosome 16 at q24.3 (arr[GRCH37]16q24.3(89851577_89897040)x0) into FANCA gene including the first exons of the gene. After the baby was born the Array CGH was processed again and same homozygote deletion was observed.
Materials and Methods
Patient
A 34 years old healthy female, was pregnant with a gestational age of 33 weeks and fetus with Encrphalocele-limbs malformation and cystic hygroma seen during her ultrasound scan. After the baby was born they processed the microarray procedure using the peripheral blood sample to confirm the result with the amniotic fluid diagnosis.
Processing the sample
Amniotic fluid harvesting
The amniotic fluid was processed by centrifuging at 2200 RPM for 10 minutes and remove the supernatant leaving around 1ml of the amniotic fluid above the pellet. Then it’s treated with Trypsin-EDTA 0.25% for 17 minutes in an incubator at 37 degrees and then spinning it for 10 minutes at 2200RPM. The supernatant was discarded leaving 1 ml above the pellet and then add 10 ml of pre warm KCL for 17 minutes for incubation. After the incubation period is done 0.5ml of Fixative which is a combination of methanol/acetic acid (3:1) is added. Then spinning again at the same RPM and then discarding the supernatant and add 6ml of fixative
DNA extraction of Amniotic fluid
The amniotic fluid is extracted manually using Qaigen DNA extraction kit. The quantity and qualityof the DNA is analyzed by using Nanodrop.
DNA Extraction of peripheral blood sample
The baby’s peripheral blood sample was used to extract DNA by using an automatic DNA extraction machine EZ1 and EZ1 DNA extraction kit. The quality and quantity of the DNA was identified by using Nanodrop.
Fluorescent in situ hybridisation (FISH)
The first test done was FISH for aneuploidy. The slide making is processed by doing 2 slides and aged at 60 °C overnight. These slides need to be pre-treated with 2X SSC for 1 hour and then for 13minutes in pepsin for digestion at 37 °C. Later these slides are process for 5 minutes in each coplin jar containing PBS and Formaldehyde solution. Dehydrate the slides using 70%, 80% and 100% ethanoland FISH is performed according to the manufacturer instructions using Anuevison probes LSI 13SG/21SO and LSI 18SA/XSG/YSO, Vysis. 10 µl of probe mixture is applied to the pre-treated slides and by using Thermobrite which runs a program for co-denaturation for 5 minutes at 72 °C andovernight hybridisation at 37 °C. The next day slides were washed for 5 minutes each in a combinationof 25ml formamide and 2X SSC solution, 2X SSC and then in in 4X SSC with tween 20. Then dehydrate the slides and then mounted with 10µl of DAPI (4, 6-diamidino-2-phenyllindole, Vysis®) and analysed using a cytovision FISH station (Cytovision, APPLIED IMAGING®).
Comparative Genomic Hybridization
Agilent Oligonucleotide array was used according to the manufacturer’s instruction “Agilent SurePrint G3 human CGH+SNP 4x180K”. Patients DNA as well as a reference DNA were digested with Rsal and Alul. Each digested DNA product was labelled with random primer using either Cy5-dUTP or Cy3- dUTP. Hybridisation was performed using a 180K microarrays. Array was scanned using the Agilent DNA micro-array scanner G2600D and analyzed with feature extraction 12.0.3 software (DLRS< 0.20). Results were interpreted with Agilent Cytogeneomics 4.0.3.12 software. A copy number variations was considered if at least three contagious oligonucleotides presented an abnormal by log ratio >+0,58 or <-1. Results were compared to Data recorded in the Database of genomic variants. This technology does not detect balanced rearrangements and may not detect low mosaicism.
Results
The FISH result for the amniotic fluid was normal and chromosome Y was detected in the process indicating a male fetus. On the other hand, Array CGH CGH analysis of both amniotic fluid as well as the peripheral blood from the baby showed a pathogenic abnormal result with homozygote deletion of 45Kb on the long arm of chromosome 16 at q24.3 (arr[GRCH37]16q24.3(89851577_89897040)x0) into FANCA gene including the first exons of the gene. Deletion of the FANCA gene have been extensively reported from exon 1 to exon 18, however, the homozygote deletion is never reported
before. The deletion result from recombinationbetween very distant Alu repeats and involve 2 adjacent genes SPIRE2 and TCF25, which heterozygous deletion would not cause dominant inherited disease, however the homozygote deletion give more severe phenotype as our case, the patient died at the age of one year and 4months. In addition, our CNV result is not detected by WES.
Discussion
The FANCA gene provides instructions for making a protein that is involved in a cell process knownas the Fanconi anemia (FA) pathway and mutations in the FANCA gene lead to a non-functional FAcore complex, which disrupts the entire FA pathway. Mutations in the FANCA gene are responsiblefor 60 to 70 percent of all cases of Fanconi anemia. These mutations change single DNA building blocks (nucleotides) or insert or delete pieces of DNA in the FANCA gene. Some mutations allow production of a FANCA protein that has some residual function; other mutations prevent theproduction of any FANCA protein. Mutations that prevent all protein production usually lead to a shortage of blood cells at an earlier age and increase the risk of developing cancer of the blood- forming cells (leukemia) as compared to mutations that allow for some FANCA protein production. The cytogenetics location for this particular gene is located on Chromosome 16q24.3.
However in our patient a pathogenic homozygotic deletion of 45 Kb on the long arm of chromosome 16 at q24.3 (arr[GRCH37]16q24.3(89851577_89897040)x0) into FANCA gene including the first exons of the gene. Deletion of the FANCA gene have been extensively reported from exon 1 to exon18, however, the homozygote deletion is never reported before. The deletion result from recombination between very distant Alu repeats and involve 2 adjacent genes SPIRE2 and TCF25, which heterozygous deletion would not cause dominant inherited disease, however the homozygote deletion give more severe phenotype as our case, the patient death at the age of one year and 4 months.In addition, our CNV result is not detected by WES.
Levran et al. (1998) reported the occurrence of Alu-mediated genomic deletions in the FANCA gene.Two different deletions of 1.2 kb and 1.9 kb were found in patients with Fanconi anemia. Both deletions included exons 16 and 17 and removed a 156- bp segment from the transcript, causing a shorter in-frame message. Introns 15 and 17 are rich in partial and complete Alu repeats. Sequence analysis of the deletion showed that the 5-prime breakpoints occurred at different sites in the same Alu element in intron 15, while the 3-prime breakpoints were located in different Alu repeats in intron 17. Numerous Alu repeats are present in the FACA gene, suggesting that Alu- mediated recombinationmay be an important mechanism for the generation of Fanconi anemia-producing mutations.
Conclusion
In conclusion, array-CGH analysis of our patient allowed us to detect a homozygote deletion of 45kb. This diagnosis authoritative to establish a precise phenotype genotype correlation and to provide genetic counselling to the couple. The risk of having children with deletion is increased. Fortunately, array CGH test could be offered to both parents and for future pregnancies can be can be checked using amniotic fluid.
Acknowledgements
We thank the family member for their continued interest and corporation
References
- Gillentine MA, Berry LN, Goin-Kochel RP, Ali MA, Ge J, et al. (2017) Hydrogen gas ameliorates oxidative stress in early brain injury after subarachnoid hemorrhage in rats. J Autism Dev Disord 47: 549-62.
- Chandrasekharappa SC, Lach FP, Kimble DC, Kamat A, Teer JK, et al. (2013). Massively parallel sequencing, aCGH, and RNA- Seq technologies provide a comprehensive molecular diagnosis of Fanconi anemia. Blood 121: e138-e148.
Artcle Information
Review Article
Received Date: June 13, 2021
Accepted Date: July 15, 2021
Published Date: July 17, 2021
Journal of Pediatric Care and Neonatology
Volume 1 | Issue 1
Citation
Tabassum T, Ben Abdallah Bouhjar I, AL Onazi HE, Babic I, Alhashem A, et al. (2021) Case Report: Array CGH Analysis Shows Pathogenic Homozygote Deletion of FANCA Gene Detected in a New Born with Multiple Congenital Abnormalities. J Pediatr Care Neonatol 1: 1-6
Copyright
©2021 Ben Abdallah Bouhjar I. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.

doi: jpcn.2022.1.1-6