

Review Article
Volume-1 Issue-1, 2021
Acute Type 2 Respiratory Failure from Hypokalemic Respiratory Muscle Paralysis Secondary to Distal (Type1) Renal Tubular Acidosis due to Primary Sjogren’s Syndrome
Received Date: January 23, 2022
Accepted Date: March 05, 2022
Published Date: March 07, 2022c
Journal Information
Abstract
Tubulointerstitial nephritis (TIN) is the main renal involvement associated with primary Sjogren syndrome (pSS). TIN can manifest as distal renal tubular acidosis (RTA), nephrogenic diabetes insipidus, proximal tubular dysfunction, and others. Distal renal tubular acidosis (RTA) may develop in a large population of patients with Sjogren’s syndrome (SS), but most of the subjects are asymptomatic. We report a case of 37 year old woman who presented with acute quadriparesis with acute hypercapnic respiratory failure. She had history of recurrent episodes of limb weakness which was attributable to hypokalemia and initially labelled as hypokalemic periodic paralysis. Lateron she was found to have metabolic acidosis rather than alkalosis which pointed towards the diagnosisof renal tubular acidosis (RTA) in the absence of apparent gastrointestinal tract loss. Once the diagnosisof RTA was established, an attempt to search the aetiology revealed that she was having primary Sjogren’s syndrome (pSS) though she did not have any symptom at the time of diagnosis. She wasfound positive for ANA, anti-SSA and anti-SSB. Schirmer test confirmed presence of dry eye. A concomitant existence ofautoimmune hypothyroidism was a noteworthy association. Continuous potassium replacementproduced rapid and complete recovery from quadriplegia and respiratoryfailure without requirement for mechanical ventilation.Presentation of this case reminds the importance of vigilancewhile managing a case of recurrent hypokalemia which might be a rare presenting feature of pSS.
Key words
Distal Renal Tubular Acidosis; Primary Sjogren’s Syndrome, Quadriparesis; Hypercapnic Respiratory Failure
Finding |
Type 1 RTA |
Type 2 RTA |
Type 4 RTA |
Normal anion gap |
Yes |
Yes |
Yes |
Minimum urine pH |
>5.5 |
<5.5 |
<5.5 |
% filtered bicarbonate |
<10 |
>15 |
<10 |
Serum potassium |
Low |
Low |
High |
Stones/ |
Yes |
No |
No |
Daily acid excretion |
Low |
Normal |
Low |
Urine anion gap |
Positive |
Positive |
Positive |
Daily bicarbonate |
<4 mmol/kg |
>4 mmol/kg |
<4 mmol/kg |
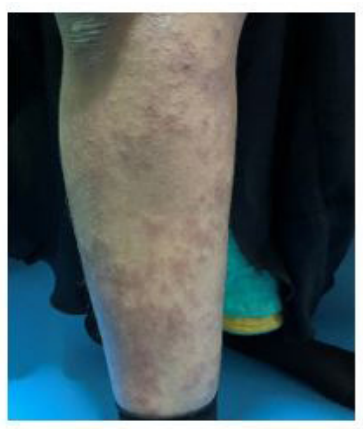 |
| Figure 1: Showing palpable vasculitic rash over lower limb |
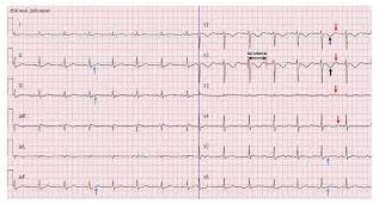 |
| Figure 2: Electrocardiogram showing sinus bradycardia, diffuse ST depression, T inversion (black arrow), U wave (red arrow) in precordial limbs and prolonged PR interval |
 |
| Figure 3:Positive Schirmer test showing 1 mm wet on filter paper after 5 minutes |
Introduction
Renal tubular acidosis (RTA) is a group of disorders in which renal excretion of acids is reduced, out of proportion to any reduction in glomerular filtration rate. RTAis characterized by hyperchloremic metabolic acidosis with a normal serum anion gap. There are multiple forms of RTA, depending on which aspect of renal acid handling has been affected (Table 1) [1]. In Type 1 or distal RTA (dRTA), the distal nephron does not lower urine pH normally because the collecting ducts permit excessive back diffusion of H+ from lumen to blood. Urinary concentration and potassium conservation also tend to be impaired, so polyuria and polydipsia occur. With the stress of an intercurrent illness, acidosis and hypokalemia can be life-threatening1. Acute hypokalemic paralysis is an uncommon cause of acute muscleweakness, and the reported cases of quadriplegia and acute respiratoryfailure secondary to hypokalemia are rare [2,3,4,5]. We report a case of dRTA who presented with hypokalemic muscular paralysis with acute hypercapnic respiratory failure who did not require ventilation because of fast respiratory function improvement by appropriate treatment with rapid potassium infusion.
Case Report
A 37 years old pleasant, married, muslim, Bangladeshi housewife, known case of hypothyroidism (on 100 mcgm of levothyroxine daily) for 16 years, not known to have any other co morbidities presented to us with the complaints of sudden weakness of both upper and lower limbs for 1 day, shortness of breath for 6 hours and unable to speak for 2 hours. Weakness was sudden in onset and increasing and became so severe that she was unable to move without the help of others and her weakness progressed to the point that she was completely bed ridden. All the proximal and distal muscles and both upper and lower limbs were equally involved but there was no muscle pain. Weakness started in both upper and lower limbs simultaneously. On query her attendant informed that she developed her symptoms after taking heavy meal. There is no history of trauma, pain in the neck or dysphagia, sensory complaints like paresthesia. She also developed breathlessness for 6 hours which was sudden and progressively increasing but not associated with chest pain, sputum or haemoptysis. For last two hours she was unable to speak but can understand speech and can follow commands like closing her eyes. She denied any history of fever, headache, visual complaints in the form of blurring of vision, diplopia, convulsion, urinary and fecal incontinence, recent vaccination. There was also no history of recent previous infection like respiratory tract infection, urinary tract infection or diarrhea. Patient’s attendant also complained of occasional rash about which they could not elaborate. She has history of similar type of attack about 1 year back which was managed conservatively in a local hospital. There was no similar type of illness in her family. She has three children and all are in good health. She had no history of abortion, no bad obstetric history and all of her pregnancy was uneventful.
On physical examination, she was conscious, oriented, but a bit drowsy with GCS 14/15 and had areflexic quadriplegia. Power was graded 0/5 in the legs and in thearms. Plantar responses were absent bilaterally.All modalities of sensation are intact, sings of meningeal irritation were absent. Cerebellar signs, stance and gait could not be evaluated. She had palpable rash over both lower limbs (figure 1), buttocks and over both palm. Her pulse was 60 beats/min, blood pressure 110/70 mmHg, respiratory rate 30 breath/min. Herchest was clear on auscultation, oxygen saturation was 97% on 5 L of oxygen. The rest of physical exam was unremarkable.
On initial laboratory investigations, complete blood count, random blood sugar, SGPT, serum creatinine, CPK, chest x ray revealed normal results except ESR was 68 mmin 1st hour . Serum Electrolyte showed sodium 138 mmol/L, potassium 1.7 mmol/L, chloride 111 mmol/L, bicarbonate 16.5 mmol/L. Anion gap was normal(12.2 mmol/L).Urinary potassium was elevated with 101mmol/L. TSH, serum calcium, serum magnesium level were normal. Electrocardiogram showed sinus bradycardia, diffuse ST depression, T inversion, U wave in precordial limbs and prolonged PR interval. (Figure 2)
Arterial blood gas (ABG) analysis revealed mixed respiratory and metabolic acidosis with hypercapnic respiratory failure (pH 7.03, PO2-69 mm, PCO2- 64 mm, HCO3-16 mmol/L). She was started treatment with intravenous sodium bi carbonate along with intravenous and oral potassium chloride and showed dramatic clinical improvement in 1 day. On next day, her muscle power improved to 4/5 in both upper and lower limbs, respiratory rate was 16 breaths/min and can speak well. SaO2 was 96% in room air. Repeat ABG in room air revealed resolved hypercapnic respiratory failure (pH 7.40, PO2-88 mm, PCO2- 30 mm, HCO3-14 mmol/L). Repeat serum electrolyte revealed sodium 147 mmol/L, potassium 2.9 mmol/L, chloride 109 mmol/L and bicarbonate 18 mmol/L. Despite of correction of respiratory acidosis her metabolic acidosis persisted.Urine routine examination showed pH 7.2, trace proteinuria without any RBC, pus cell or any casts. So type 1 or distal renal tubular acidosis (dRTA) was suspected and for further evaluation of underlying cause for dRTA, on query, she revealed that she has been suffering from occasional rash for last 5 years which was more prominent in her legs and buttock area and also involved her palm sparing her trunk and face. Rashes care initially itchy and then became painful. She developed these rashes occasionally for last 5 years and resolved spontaneously. She denied any history of joint pain, oral ulcer, muscle pain, hair fall, and weight loss.She also complained of dryness of mouth for last 15 years, thatwas increasing and became so severe that she cannot swallow her food without water. She also developed dry eyes for same duration and there is no tear during her emotional event like crying. Bedside Schirmer’s test was done and found positive (1 mm wet on filter paper in 5 minutes- figure 3). She also complaints of bluish discoloration of her fingers and pain on cold exposure and she has been suffering from fatigue and weakness for last 15 years and dry cough and worsening shortness of breath on exertion for last 2 years.With these symptoms, she visited a number of physicians including ophthalmologists and was managed symptomatically without any improvement.
Further investigations revealed: ANA was strongly positive on Hep2 cell with fine speckled pattern. Anti dS DNA, lupus anti coagulant, anti cardiolipin antibody was negative. ENA profile showed positive anti SSA(Ro) and anti SSB(La).RA test was positive in high titer, Serum total protein was 77 gm/L, albumin 35 gm/L and globulin 42 gm/L, albumin globulin ratio was 1: 1.42. Anti TPO was > 1300 U/ml(positive >60 U/ml).C4 level was reduced to 0.1 g/L(normal 0.15-0.45 g/L). Protein Electrophoresis showed polyclonal hypergammaglobulinemia with prominent IgG and IgM. Pulmonary function tests showed mild restrictive defect as evidenced by low FEV1(2.02 L) and low FVC(2.05L) with normal FEV1/FVC(94.5%). DLCO was moderately reduced with 55% of predicted value. KCO was normal suggestive of parenchymal restrictive disease. Patient refused for lip biopsy. So final diagnosis of recurrent hypokalemic paralysis due to distal (type 1) renal tubular acidosis secondary to primary Sjogren’s syndrome with type 3 cryoglobulinemic vasculitis with autoimmune hypothyroidismwas made. She was discharged with sodium bicarbonate, potassium citrate, hydoxychloroquine (400 mg daily) and artificial tear. After 1 month, on outpatient door follow up she was doing well.
Discussion
Approximately, 98% of total body potassium is located intracellularly. Sixty percent of intracellular potassium is within skeletal muscle, which may explain the predominance of muscular symptoms in disorders producing hypokalemia [6,7]. Although, the effects of hypokalemia are multisystemic, potassium deficiency most seriously affects the neuromuscular system, and symptoms related to hypokalemia are typically muscular . Serum potassium concentrations of 3.0 to 3.5 mmoU1 may be associated with mild muscle weakness and myalgia. Serum potassium concentrations of 2,5 to 3.0 mmol/L are associated with the development of clinically significant muscle weakness. The muscle weakness generally is limited to the limbs and limb girdles. The respiratory or cranial musculature are rarely involved and characteristically spared in muscle weakness secondary to hypokalemia. When severe, hypokalemia can impair respiratory function leading to hypoventilation, and acute respiratory failure [2,3,4,5,8,9]. Hypokalemic muscle weakness may simulate Guillain -Barre syndrome [10,11,12]. When the serum potassium level fallsbelow 2.5 mmol/L, rhabdomyolysis may occur[7,13,14]. The lowest serum potassium level reported was 1.1 mmol./L this was associated with areflexic quadriplegia, coma, and acute respiratory failure managed with IPPV [8].
Hypokalemia is a common clinical problem, and is the most frequent electrolyte disorder encountered in clinical practice. The major causes ofhypokalemia are: Inadequate potassium intake, excessive skin, renal(diuretics, adrenal insufficiency, salt losing nephropathy), orgastrointestinal losses(diarrhoea, vomiting), and increased entry into cells by a variety ofmechanisms including increased availability of insulin, elevated beta adrenergicactivity and elevation in extracellular pH (metabolic orrespiratory alkalosis).
RTA is characterized by hyperchloremic metabolic acidosis with a normal serum anion gap. Thereare multiple forms of RTA, depending on which aspect of renal acid handling has been affected (Table 1).
The presence of severe hypokalemia (K+ 1.7mmol/L) with normal anion gap metabolic acidosis and urinary pH >5.5 suggested the diagnosis of Type 1 dRTA. Although quadriplegia is a wellknown complication of severe hypokalemia, acute respiratory failure due to dRTA is rarely reported [15]. A thorough search of the available literature (Medline) revealed only six cases of dRTA leading to hypokalemic respiratory failure requiring mechanical ventilation [16-21].
Ascending paralysis of the extremities suggested Guillain-Barré syndrome, however the presence of hypokalemia, metabolic acidosis and an alkaline urine confirmed the diagnosis, and lumbar puncture was not performed. Moreover, there was a significant improvement in muscle power after K+ supplementation.
The pathogenesis of classic hypokalemic dRTA is not yet clear. The hallmark is the inability to acidify the urine to pH < 5.5; there is no impairment in bicarbonate reabsorption in proximal tubules1. In the majority of patients, there is a defect in H+ secretion by the H+-K+-ATPase pump in tubular cells which causes hypokalemia, decrease in NH4+ excretion, hyperchloaemic metabolic acidosis and volume depletion. Occasionally, strikingly severe hypokalemia, metabolic acidosis and hypocalcaemia may require immediate therapy. This constellation of findings in an emergency setting has been labelled as a “crisis of dRTA” [22]. Clinical presentation in our patient was also quite catastrophic, with quadriparesis, acute respiratory failure which could have proved fatal without prompt and effective management.
Chronic positive acid balance causes Ca2+, Mg2+ and PO43- wasting and there is an increased incidence of nephrocalcinosis. A prominent feature of dRTA is abnormal Ca2+ metabolism leading to musculoskeletalcomplaints. Hypercalciuria, alkaline urine and low levels of urinary citrate result in calcium phosphate stones [22].
The majority of patients with dRTA have it in association with a systemic illness such as Sjögren’s syndrome, hypergammaglobulinaemia, chronic active hepatitis (CAH) or lupus. The frequency of dRTA in Sjögren’s syndrome has been reported to be 25 to 40%22. Ohtani et al and Poux et al have described hypokalemic quadriplegia and respiratory arrest due to dRTA in patients with primary Sjögren’s syndrome [18,19]. Koul and Saleem reported a case of CAH with dRTA which presented with hypokalemic muscular paralysis requiring respiratory assistance [21].
The diagnostic criteria of Sjogren's syndrome were satisfied by the findings of xerostomia, high titer of anti SSA and anti SSB antibodies. The frequency of dRTA in Sjogren’s syndrome has been reported to be about25-40%. [23,24]. There are many case reports of Sjogren’s syndrome associated with hypokalemic quadriplegia [25-31]. Tsuboi et al described that the periodic paralysis was observed in almost 40% of Sjogrensyndrome cases associated with dRTA31. However only three cases were reported that respiratory arrest depends on severe hypokalemia associated with renal tubular acidosis due to various causes [32-34] and there is only one case reportdescribing respiratory failure due to Sjogren’s syndrome [34]. There is no significant difference at the level of serum potassium between respiratory arrest group and quadriplegia alone group (1.0-2.8 mmol/L). These results suggests that the progress for respiratory arrest may be may be influenced by interindividual differences in the sensitivity towards hypokalemia (and presumably also transmembrane K gradient) of respiratory muscles. Even patients with hypokalemia< 1.7 mEq/l were liable to develop respiratory arrest. The above cases, including our own patient, were all female, and showed good prognosis with immediate respiratory support and potassium supplementation. However, Nimmannit etal. [35] described fatal cases of hypokalemic respiratory failure and ventricular fibrillation due to endemic RTA inThailand. This condition occurred in otherwise healthy young males and sometimes resulted in nocturnal death which is different from the present case.
Treatment of dRTA involves correction of metabolic acidosis by administration of alkali in an amount sufficient to neutralize the production of diet-derived acids. This is usually equal to 1-3 mmol/kg/.day of sodium bicarbonate in adults. In children, a large amount of bicarbonate must be administered to correct acidosis and maintain normal growth. Sustained correction of metabolic acidosis usually suppresses renal Na+ and K+ wasting with attendant correction of extracellular fluid volume and hypokalemia. Therefore in most patients with dRTA, K+ supplementation is not necessary. However, if the patient presents with hypokalemic respiratory paralysis, the K+ deficit should be replaced without delay before initiating bicarbonate therapy [22], because if bicarbonate is given alone it may further worsen the already existing hypokalemia. The rate of infusion of KCl should be up to a maximum of 0.25 mmol/kg/.hr. These patients are often volume depleted and treatment should include correction of the fluid deficit if necessary guided by central venous pressure monitoring.
It is well known that hypokalemia can cause muscle weakness and respiratory failure, but is probably under-appreciated. A prompt diagnosis and treatment of hypokalemia is warranted to prevent respiratory failure and the requirement for mechanical ventilation, and to prevent other complications, mainly cardiac.
Conclusion
This case report highlights the fact that the dRTA with severe hypokalemia should be kept in mind in any patient presenting with respiratory failure. Secondly, all patients with dRTA must be investigated for any associated systemic illnesses and put on lifelong alkalinizing agents. In pSS, RTA is commonly asymptomatic. However, if such a state persists, patient may develop quadriplegia. Furthermore, if adequate treatment is not received, muscle paralysis may progress to respiratory arrest. Although respiratory arrest associated with Sjogren’s syndrome is very rare, this complication is very severe and can be fatal. It is important to pay attention to the occurrence of severe hypokalemia with metabolic acidosis and provide adequate treatment for this combination in patient with pSS.
Conflict of Interest:
None declared.
References
- Asplin JR, Coe FR (2001) Hereditary Tubular Disorders. In: Braunwald E, Fauci AS, Kasper DL et al, eds. Harrison’s Principles of Internal Medicine. 15th Ed. New York: McGraw-Hill 1598-606.
- Mutlu Gm, Factor P (2002) Acute-onset quadriplegia, respiratory failure, and ventricular tachycardia in a 21-year-old man following a soccer match. Chest 121: 2036-9.
- Da Vies Rg, Gemmell L (2001) Severe hypokalaemia causing acute respiratory failure. Anesthesia 5: 694-5.
- Puddy Br (2000) Acute respiratory failure secondary to severe hypokalaemia. Anaesthesia; 55: 1221-2.
- Dunn Sr, Famsworth Ta, Karunaratne Wu (1999) Hypokalaemic, hyperchloraemic metabolic acidosis requiring ventilation. Anaesthesia; 54: 566-7.
- Knochel JP (1982) Neuromuscular manifestations of electrolyte disorders. Am J Med; 72: 521-35.
- Jacke Riggs (2002) Neurologic manifestations of electrolyte disturbance. Neural Clin 20: 227-39.
- Latiere M, Dumont Jc, Olmer M, Francois G (1989) Hypokalaemic quadriplegia and coma associated with renal tubular acidosis. Ann Fr Anesth Reanim; 8: 133-6.
- Fogliani J, Giraud E, Henriquet D, Maitrasse B (1993) Voluntary barium poisoning. Ann Fr Anesth Reanim; 12: 508-11.
- Koley Tk, Goyal Ak, Gupta Md (2001) Barium carbonate poisoning mimicking Guillain-Barre syndrome. J Assac Physicians India 49: 656-7.
- Warner Tt, Mossman S, Murray NM (1993) Hypokalaemia mimicking Guillain-Barre syndrome. J Neural Neurosurg Psychiatry; 56: 1134-5.
- Royston A, Prout Bj (1976) Carbenoxolone-induced hypokalemia simulating Guillain-Barre syndrome. BMJ; 2: 150-1.
- Campion Ds, Arias Jm, Carter NW (1972) Rhabdomyolysis and myoglobinuria. Association with hypokalemia of renal tubular acidosis. J Am Med Assoc; 220: 967-9.
- Kao Kc, Tsai Yh, Lin Mc, Huang Cc, Tsao Cy, et al. (2000) Hypokalemic muscular paralysis causing acute respiratory failure due to rhabdomyolysis with renal tubular acidosis in a chronic glue sniffer. J Toxical Clin Toxical 38: 679-81.
- Ahuja GK, Mittal VK (1981) Periodic paralysis with renal tubular acidosis. Neurol India 30: 188-91.
- Tyagi A, Rautela RS, Bhattacharya A (2002) Renal tubular acidosis presenting with respiratory arrest. Indian J Pediatr 69: 197.
- Fujimoto T, Shiiki H, Takahi Y, Dohi K (2001) Primary Sjögren’s syndrome presenting as hypokalemic periodic paralysis and respiratory arrest. Clin Rheumatol 20: 365-8.
- Ohtani H, Imai H, Kodama T et al. (1999) Severe hypokalemia and respiratory arrest due to renal tubular acidosis in a patient with Sjögren’s syndrome. Nephrol Dial Transplant 14: 2201-3.
- Poux JM, Peyronnet P, Le Meur Y et al. (1992) Hypokalemic quadriplegia and respiratory arrest revealing primary Sjögren’s syndrome. Clin Nephrol 37:189-91.
- Alvarez J, Low RB (1988) Acute respiratory arrest due to hypokalemia. Ann Emerg Med 17: 288-9.
- Koul PA, Saleem SM (1992) Chronic active hepatitis with renal tubular acidosis presenting as hypokalemic periodic paralysis with respiratory failure. Acta Paediatr 81: 568-9.
- Du Bose TD Jr, Cogan MG, Rector FC Jr. (1996) Acid Base Disorders. In: Brenner BM, ed. Brenner &Rector’s The Kidney.5th Ed. Philadelphia: WB Saunders Company 929-98.
- Shearn MA,Tu WH (1968) Latent renal tubular acidosis in Sjogren’s syndrome. Ann Rheum Dis 27: 27-32
- Siamopoulos KC, Mavridis AK, Elisa fM, Drosos AA, Moutsopoulos HM (1986) Kidney involvement in primary Sjogren’s syndrome. Scand J Rheumatol1986[Suppl] 61: 156-60
- Christensen KS (1985) Hypokalemic paralysis in Sjo¨gren’s syndrome secondary to renal tubular acidosis. Scand J Rheumatol 14: 58-60
- Nagashima K (1996) A patient with Sjo¨gren syndrome with central pontine myelinolysis and hypokalemic myopathy. ClinNeurol 36: 1240-4
- Pun KK, Wong CK, Tsui EYL, Tam SCF, Kung AWC, et al. (1989) Hypokalemic periodic paralysis due to the Sjogren syndrome in Chinese patients. Ann Intern Med 110: 405-6
- Chang YC, Huang CC, Chiou YY, Yu CY (1995) Renal tubular acidosis complicated with hypokalemic periodic paralysis. Pediatr Neurol 13: 52-4
- Raskin RJ, Tesar JT, Lawless OJ (1981) Hypokalemic periodic paralysis in Sjo¨gren’s syndrome. Arch Intern Med 141: 1671-3
- Hattori N, Hino M, Ishihara T, Moridera K, Ikekubo K, et al. (1992) Hypokalemic paralysis associated with distal renal tubularacidosis. Intern Med 31: 662-5
- Tsuboi Y, Niijima K, Jyoshita Y, Yoshida M, Muto S, et al. (1990) A case of renal tubular acidosis typeI RTA with Sjo¨gren’s syndrome, hypokalemic periodic paralysis, and selective hypoaldosteronism. SaishinIgaku 45: 610-7(in Japanese , English abstract)
- Bridi GS, Falcon PW, Brackett NC, Still WJS, Sporn IN (1972) Glomerulonephritis and renal tubular acidosis in a case of chronic active hepatitis with hyperimmunoglobulinemia. Am JMed 52: 267-78
- KoulPA, Saleen SM (1992) Chronic active hepatitis with renal tubular acidosis presenting ashypokalemic periodic paralysis with respiratory failure. Acta Paediatr 81: 568-9
- Poux JM, Peyronnet P, Le Meur Y, Favereau JP, Charmes JP, et al. (1992) Hypokalemic quadriplegia and respiratory arrest revealing primary Sjo¨gren’s syndrome. Clin Nephrol 37: 189-91
- Nimmannit S, Malasit P, Chaovaku, Susaengrat W, Vasuvattakul S, et al. (1991) Pathogenesis of sudden unexplained nocturnal death(laitai) and endemic distal renal tubular acidosis. Lancet 338: 930-2.
Artcle Information
Review Article
Received Date: January 23, 2022
Accepted Date: March 05, 2022
Published Date: March 07, 2022
Journal of Rheumatology and Musculoskeletal Diseases
Volume 1 | Issue 1
Citation
Richmond R Gomes, Habiba Akhter (2022) Acute Type 2 Respiratory Failure from Hypokalemic Respiratory Muscle Paralysis Secondary to Distal (Type1) Renal Tubular Acidosis due to Primary Sjogren’s Syndrome. J Rheumatol and Musculoskeletal Dis 1(1):101
Copyright
©2022 Richmond R Gomes. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.

doi: jrmd.2022.1.101